Syn. Lipidthesaurismosen
Gemeinsam ist eine Störung im Abbau von Lipiden auf der Basis des Sphingosins. Diese Lipide kommen v.a. im ZNS vor. Vor allem handelt es sich im Cerebroside und Ganglioside sowie Sphingomyelin.
Grund für die Abbaustörung ist ein genetischer Defekt von lysosomalen Enzymen. Bestimmte Lipide akkumulieren daher in den Lysosomen und führen schließlich zu Zellnekrosen und Organfehlfunktionen.
Wichtige Erkrankungen:
Syn: GM2 -Gangliosidose
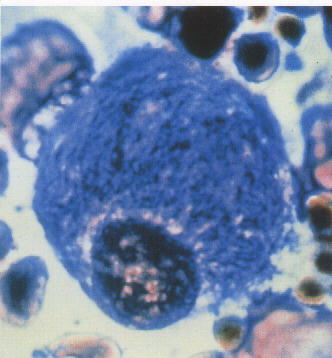
Speicherung von Gangliosid GM2 in den Lysosomen ==> Amaurotische Idiotie (Erblindung, geistige Entwicklungsstörungen)
In der späten Säuglingszeit zunehmendes Erbrechen, rezidivierende Pneumonien, progressive Makrozephalie nach dem 16. Lebensmonat infolge zerebraler Gliose. Lipidose kortikaler, autonomer und rektaler Mukosa-Neuronen mit balloniertem Zytoplasma und peripher abgedrängtem Zellkern. Zentrale Demyelinisierung, kortikale Gliose. Keine pathologischen Veränderungen viszeraler Organe bei Morbus Tay-Sachs, geringe Hepatosplenomegalie bei Morbus Sandhoff.
Biochemisch ab Geburt. Klinik: Früheste Symptome zwischen 3.-6. Lebensmonat.
Die Tay-Sachs-Erkrankung ist ein autosomal-rezessiver Hexosaminidase-A-Mangel. Die Hexosaminidase A besteht aus zwei nicht identischen Untereinheiten, der Alphakette, kodiert durch einen Locus auf den Chromosom 15, und der Betakette, codiert durch einen Locus auf dem Chromosom 5. Mutationen am Alpha-Locus des Chromosom 15 führen zur Tay-Sachs-Erkrankung bzw. der juvenilen oder adulten Verlaufsform der GM2 -Gangliosidose. Mutationen am Beta-Locus auf dem Chromosom 5 führen zum Morbus Sandhoff. Gegenwärtig kennen wir 11 Verlaufsformen der GM2-Gangliosidosen (6 Hexosaminidase-Mutanten A, 3 Hexosaminidase-B-Mutanten und 2 Aktivator-Mutanten). Genlokalisation für den M. Sandhoff auf den langen Arm des Chromosom 5, für den M. Tay-Sachs auf den langen Arm des Chromosom 15 (15q23-24).
Dazu eine sehr rührende Fallgeschichte auf http://home.arcor-online.de/bels/simon.htm Von dieser Seite stammt auch der größte Teil dieser Informationen. Danke und alle gute an die Familie Bels!
Die Speicherung von Glucocerebrosid aufgrund des autosomal-rezessiven Defekts der Glycocerebrosidase führt neben ZNS-Störungen zu einer Hepatosplenomegalie und Knochenmarksbeteiligung. Insbesondere sind Makrophagen von der Akkumulation betroffen, sie werden als Gaucher-Zellen bezeichnet.
Am häufigsten reichern sich Gaucher-Zellen in der Milz, der Leber und dem Knochenmark an. Sie können aber auch in anderen Geweben gespeichert werden, u.a. dem Lymphsystem, den Lungen, der Haut, den Augen, der Niere und - sehr selten - im Nervensystem. Ein Organ, das Gaucher-Zellen enthält, ist häufig vergrößert und zeigt Funktionsdefizite, was zu klinischen Symptomen führt, die typisch für die Krankheit sind.
Bei M. Gaucher Typ 2 und 3 (äußerst selten) ist außerdem des Nervensystem betroffen. Zusätzliche Symptome: Augenbewegungsstörungen, d.h. Schielen, das operativ nicht behoben werden kann; Epileptische Anfälle; Störungen in der Bewegungsmotorik.
autosomal-rez. Defekt der Sphingomylinase ==> Akkulation von Sphingomyelin
Hypothese: Nicht erst das Sphingomylin wird nicht abgebaut, sondern auch sein Vorläufer, das Lysosphingomyelin. Dieses inhibiert die Phosphokinase C ==> Störung der Signaltransduktion
|
Typ A |
Typ B | Typ C |
|
|
|
Synonyme: a-Hydroxylase-Mangel, Heredopathia atactica polyneuritiformis
Durch a-Hydroxylase-Mangel ==> Störung im Abbau verzweigtkettiger Fettsäuren, insbesondere der Phytansäure , die endogen nicht synthetisiert wird, gestört. Der Nachweis erfolgt daher durch den erhöhten Phytansäurespiegel im Serum.
Fakultativ bestehen Pupillenstörungen, Innenohrschwerhörigkeit, Katarakt, Anosmie, kardiale Störungen, Ichthyosis-ähnliche Hautveränderungen, Skelettdeformitäten. Der Verlauf ist schubförmig mit Teilremission und akuten Exazerbationen.
Der Krankheitsverlauf beträgt 20-30 Jahre.