Paraproteinämien
monoklonale Gammopathien
Sehr heterogene Gruppe von Erkrankungen, die Gemeinsamkeit besteht im
gehäuften Auftreten von monoklonalem Immunglobulin (meist ohne Antikörperfunktion)
im Blut oder Harn. Dieses g-Globulin wird als
Paraprotein, Myelomprotein oder M-Komponente bezeichnet.
Das Paraprotein ist das Produkt eines einzigen, proliferierende
Plasmazell-Klons.
Einteilung der Gammopathien
| primäre (maligne) |
sekundäre (benigne) |
|
|
- Karzinome nicht lymphoretikulärer Ursache
- Monozyten-Leukämie
- Chronische Hepatitis
- Chronische Entzündungen (hier allerdings häufiger oligo- oder
polyklonale Globulin-Verschiebungen)
- Rheumatoide Arthritis
- Cold-Agglutinin-Syndrom
- Immundefekte
- Morbus Gaucher (Lipidspeicherkrankheit)
- Medikamente
|
Sekundäre monoklonale Gammopathien können vorübergehend sein oder
persistieren. Jedenfalls müssen sie jahrelang kontrolliert werden, um ein
beginnendes Plasmozytom zu erkennen.
In etwa 50%-70% der Fälle werden IgG produziert, in ca. 15-25% IgA,
ebenfalls etwa 15% IgM (Makroglobulinämie, Morbus Waldenström), selten IgD, vereinzelt
IgE ohne AK-Aktivität.
Beim Bence-Jones-Plasmozytom werden ausschließlich L-Ketten produziert.
Vorkommen meist jenseits des 50.·Lj.; Männer sind häufiger betroffen, die
Ursache der Erkrankung ist unbekannt.
Symptomatik:
- oft symptomlos
- lokale (belastungsabhängige) Schmerzen u. evtl. Spontanfrakturen durch osteolytische Skelettdestruktion mit multiplen Knochendefekten
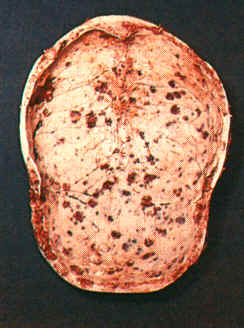
(bei M. Kahler v.·a. am Schädel (s. Abb.) u. im Bereich der Wirbelsäule
infolge Plasmazellinfiltration.
- Durch hohen Proteinanteil im Plasma ==> Hyperviskositätssyndrom (absolute
Hyperproteinämie)
- rezidivierende Infekte (Immundefekt)
- Abgeschlagenheit
- Gewichtsverlust
- Durch Ausscheidung von Leichtketten-Paraproteinen im Urin (Bence-Jones-Proteinurie in ca. 60-80% der Fälle)
==> AL-Amyloideinlagerungen in das Nierenparenchym ==>
Niereninsuffizienz
- Die fortschreitende Ausbreitung im Knochenmark führt durch Verdrängung des blutbildenden Gewebes häufig zur Anämie, evtl. zur Granulozytopenie u.
gelegentlich zu einer Thrombopenie (hämorrhag. Diathese)
- In späten Krankheitsstadien Übergang zur Plasmazellenleukämie möglich.
Diagnostik:
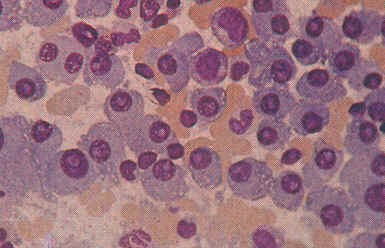
- Knochenmarkbiopsie u. histol. Nachweis einer Vermehrung von Plasmazellen (s.
Abb.)
- Nachweis einer Paraproteinurie mittels Immunelektrophorese
- Rö:.Nachweis von osteolyt. Knochendefekten
- labordiagn. stark beschleunigte BKS, Zeichen einer Niereninsuffizienz, Anämie
Therapie:
In Abhängigkeit von Stadien u. klin. Symptomatik
- Glukokortikoide u.
- Zytostatika
- Strahlentherapie bei isolierten Knochenherden
- bei pathol. Fraktur Osteosynthese
- Schmerztherapie
Prognose: mittlere Überlebenszeit unter Chemotherapie ca. 3-4 Jahre.